ChemGPS-NPweb
A tool tuned for navigation in biologically relevant chemical space
Introduction
The World Wide Web has become a central source for information, education, tools, and services that make life easier for medicinal chemists and drug discoverers. Internet technology offers an exceptional possibility to develop public tools. We have developed a web-based public tool ChemGPS-NPweb,(http://chemgps.bmc.uu.se/), for comprehensive chemical space navigation and exploration in terms of global mapping on to a consistent 8-dimensional map of structural characteristics. ChemGPS-NP [1, 2] is a principal component analysis (PCA) based global space map or a chemical global positioning system [3]. Compounds of interest or under study are positioned onto this map using interpolation in terms of PCA score prediction. The properties of the compounds together with trends and groupings can easily be interpreted from the resulting projections. In this article we review design, features, and proposed fields of application of ChemGPS-NPweb.
Technical details
General
ChemGPS-NPweb includes a number of different programs and libraries that interact with each other according to the traditional UNIX-model. Each element performs a well defined task and together they solve a more advanced problem.
The system includes three main elements: DragonX [4], for calculation of molecular descriptors, Simca-QP [5], for multivariate predictions, and the web interface (Batchelor). Further more a batch queue manager is used. This allows jobs with long run times to be submitted to the web server and scheduled for later execution by its batch queue. The programs exchange information with the web interface by storing information in the file system, which acts as the database
Job flow
When the job queue starts a job, the following things occur: the uploaded SMILES [6] strings are processed by DragonX, and the obtained data are then transformed by a Perl script that organizes the values of the 35 descriptors used by the model. These transformed results are used as indata to cgpsclt (client) that connects to cgpsd (server) to start the multivariate prediction. Subsequently Simca-QP performs the prediction via libchemgps and cgpsd sends the result back to cgpsclt, which stores the result in the database. If cgpsd (the server) is not available the prediction will instead be performed locally by cgpsstd (standalone program). Figure 1 describes how the different elements interact.
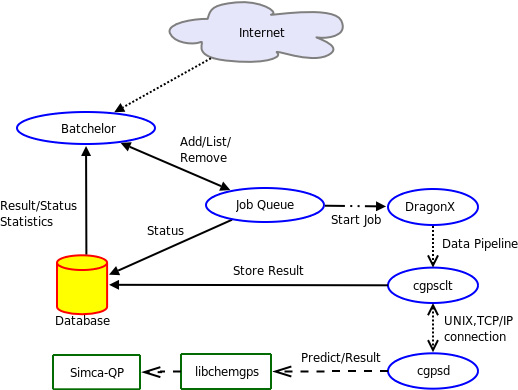
Figure 1. Flowchart describing the interaction between the different elements of ChemGPS-NPweb.
Optimizations
The extra step with client/server (cgpsclt/cgpsd) was incorporated to avoid having to load the project (reference set) for each job. As an additional benefit it also enables predictions to be performed by one or more computers on the network.
All elements (DragonX, Simca-QP, and cgpsd) are multithreaded, which becomes more and more important as the number of cores (CPUs) will increase in the future.
The web interface
The web interface (Batchelor) enables the upload of data (SMILES) and to obtain the results from the runs. The job queue can be filtered and sorted according to different criteria. Uploaded data and results are personal and can only be reached from the same computer as the job was initiated from. The information presented to the user is in part obtained from the database (results and statistics), or directly from the job queue (job status).
System information
The entire process runs at present on one single computer, a 64 bit 2 x Quad Core Xeon operating at 1.6 GHz with 4 GB RAM, and featuring a GNU/Linux operating system.
How to use ChemGPS-NPweb
The simple instruction for using ChemGPS-NPweb is as follows:
a correct SMILES-file [6] with a maximum of 500 compounds (or 1024 kB) is uploaded and submitted using the buttons ‘Browse’… and ‘Send File’ (figure 2). Any IDs in the file should be placed after (to the right of) the SMILES string. Alternatively the SMILES can be pasted in the ‘Process data’ drop box and submitted by clicking ‘Send Data’.
The resulting ChemGPS-NP 8D coordinates are obtained through ‘View results’ in the left menu. Users (submitters) can monitor the state of their submitted jobs (pending, running or finished) and later download the result from the queue view (figure 3). The coordinates (figure 4) can then be plotted using preferred software. Here we have used Grapher 2.0 distributed together with MacOS X (figure 5). From the plot it is evident that the two compounds indicated by the circle have very similar physical chemical properties as can be confirmed by the chemical structures displayed in figure 6.
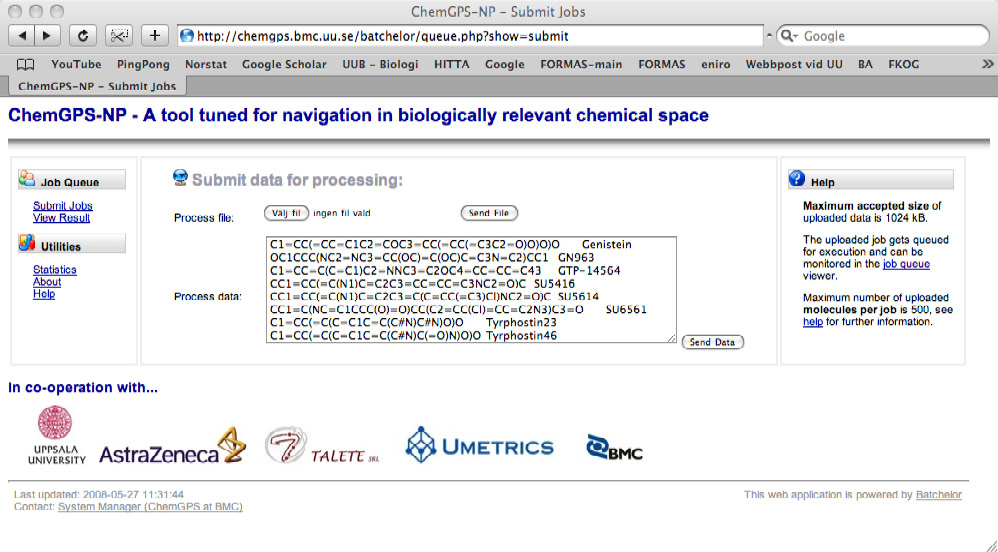
Figure 2. User upload interface of ChemGPS-NPweb.
Additionally post computational statistics are prepared based on results from each of the successive computational steps, and can be viewed by clicking ‘Statistics’ in the left menu (figure 2).
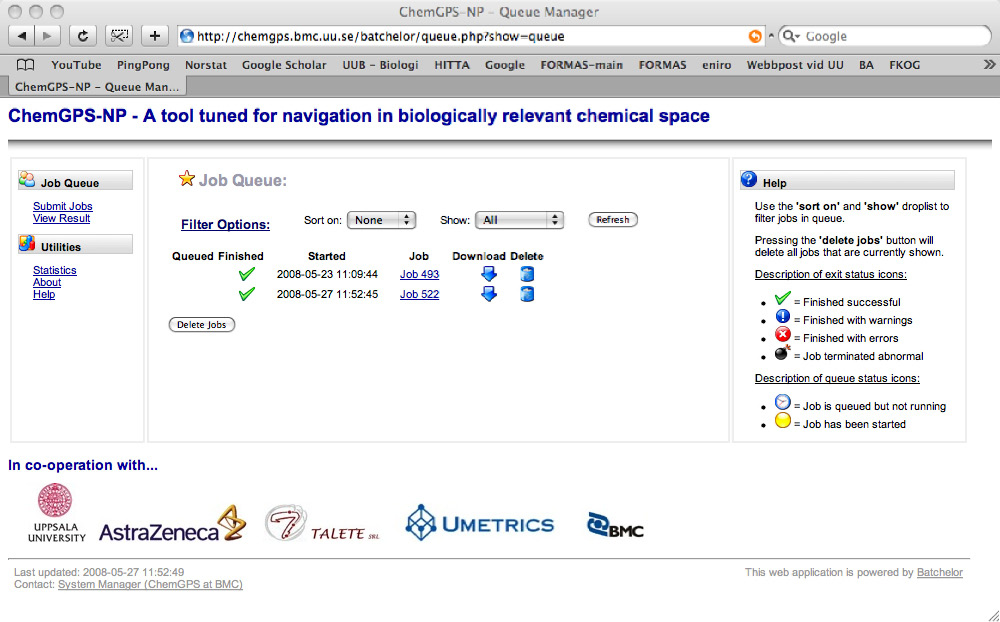
Figure 3. Queue feedback interface, allowing simple error tracing features as well as access to retrieved data both directly and as a downloadable file.
Final remarksS
The drug discovery process is today held back by increasing costs and high attrition-rates, with an overall decrease in the number of annually registered new chemical entities. Considering the immensity of chemical space, which is estimated to exceed 1060 possible compounds when only small carbon-based compounds are considered [7], it is obvious that the process of compound selection and prioritization is crucial. An efficient selection process would give a higher probability of obtaining a lead compound. ChemGPS-NP provides a framework for making compound comparison and selection more efficient, thereby increasing probability of hit generation in the search for novel bioactive molecules. The benefits of ChemGPS-NP are, in one way, comparable to the possibilities opened in molecular biology by rigorous application of the BLAST algorithms [8]. These allow, for example through web-interfaces, the research community to easily compare sections of nucleotide or amino-acid sequences for homology searching, identifying genes, or preparing datasets for phylogenetic analyses, all in huge datasets.
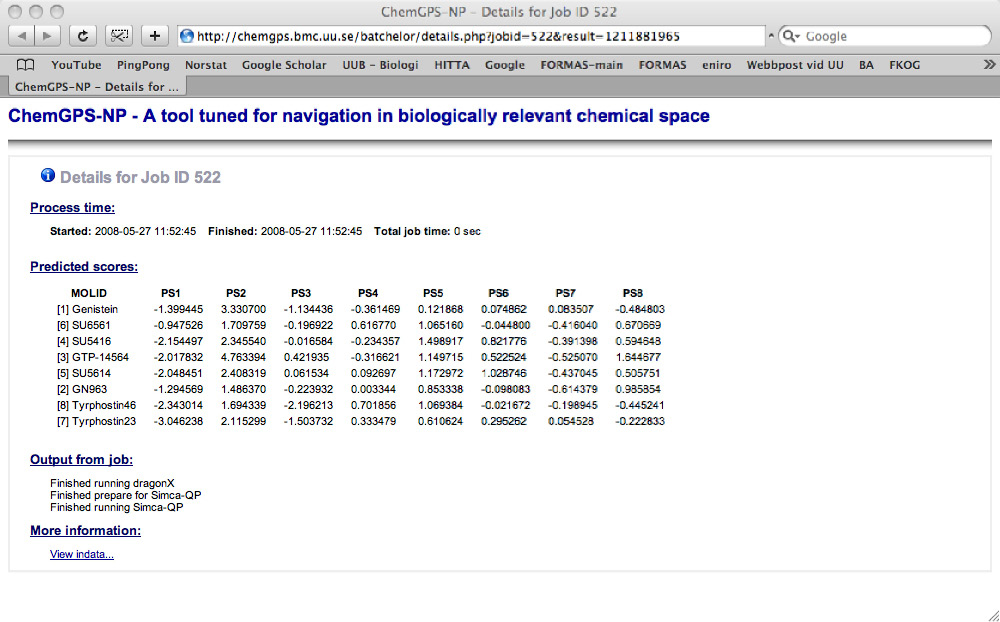
Figure 4. Resulting coordinates for submitted compounds retrieved through direct access.
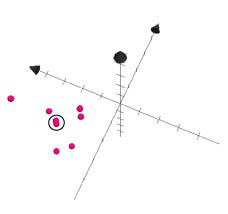
Figure 5. Retrieved data plotted using Grapher 2.0, showing first three of eight dimensions of chemical space as defined by ChemGPS-NP. From the plot it is obvious that two compounds indicated by the circle have very similar physical chemical properties.
In summary we have developed an internet tool for chemical space navigation. ChemGPS-NPweb can assist in for instance compound selection and prioritization; property description and interpretation; clustering overviews; as well as comparison and characterization of large datasets. ChemGPS-NPweb so far includes several pieces of commercial software that connect via scripts handling input, queue and output of data files. The output files can be piped into other software for post-processing, plotting and visualization.
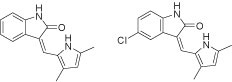
Figure 6. Molecular structures (drawn with ChemDraw Ultra 11.0.1) of the two apparently similar compounds encircled in figure 5.
Acknowledgements
Instrumental at initial stages in implementing the ChemGPS-NPweb were Thierry Kogej at AstraZeneca R&D, Mölndal, and Gustavo Gonzales-Wall and Nils-Einar Eriksson at the IT-/Computing Dept. at BMC. The authors are grateful for software support from UMETRICS and TALETE.
References
- Larsson J, Gottfries J, Bohlin L & Backlund A (2005) Expanding the ChemGPS chemical space with natural products. J Nat Prod 68: 985-991.
- Larsson J, Gottfries J, Muresan S & Backlund A (2007) ChemGPS-NP: tuned for navigation in biologically relevant chemical space. J Nat Prod 70: 789-794.
- Oprea T I & Gottfries J (2001) Chemography: the art of navigating in chemical space. J Comb Chem 3: 157-166.
- Talete srl, DragonX (Software for Molecular Descriptor Calculations). Linux version - 2007 - http://www.talete.mi.it/. Accessed May 28, 2008.
- SIMCA-QP software, Umetrics AB, Umeå, Sweden. http://www.umetrics.com/ . Accessed May 28, 2008.
- Weininger D (1988) SMILES, a chemical language and infromations system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 28: 31-36.
- Bohacek R S, McMartin C & Guida W C (1996) The art and practice of structure-based drug design: a molecular modeling perspective. Med Res Rev 16: 3-50.
- Altschul S F, Gish W, Miller W, Myers E W & Lipman D J (1990) Basic local alignment search tool. J Mol Biol 215: 403-410.